 info@naturaleyecare.com
info@naturaleyecare.com



 Home
Home


 Vision
Vision Vision
Vision



 Health
Health Health
Health Research/Services
Research/Services Pets
Pets About/Contact
About/Contact
Retinitis Pigmentosa
Treatment What it is Types Symptoms Causes Retina structure News Reviews
Retinitis pigmentosa comprises a group of inherited disorders, such as Leber's, in which the rods and cones that make up the photoreceptor cells in the retina are dysfunctional, due to genetic mutations. But about half of retinitis pigmentosa (RP) cases appear to be isolated with no previous family history.
 Next:
Nutritional support, diet, &
lifestyle tips for retinitis pigmentosa.
Next:
Nutritional support, diet, &
lifestyle tips for retinitis pigmentosa.
Retinitis pigmentosa is a progressive degenerative disorder of the photoreceptors and/or retinal pigmented epithelium (RPE) that can cause a profound loss of vision. The vast majority of patients are over 45 years old, although the symptoms of RP often appear in childhood, causing difficulty in seeing at night, and requiring longer time in adjusting from dark to light.
Retinitis pigmentosa primarily affects the side (peripheral) vision due to deterioration of the rod photoreceptors, but in later life it can affect the cones, rarely resulting in total blindness, but possibly leading to the state of “legal blindness” by age 40. Most people retain central vision with restricted side vision into their 50s. This is referred to as tunnel vision.
Genes, Photoreceptors, and Retinal Epithelium
The retinal pigment epithelium (RPE) layer lies between the Bruch's membrane and the layers of photoreceptor nerve cells, which is where light is detected and transferred to the optic nerve, and then, to the visual cortex of the brain. Not only is the RPE responsible for allowing nourishment to pass through to the photoreceptor cells, but it also protects the retina from damaging exposure and helps maintain the structural integrity of the retina.
Photoreceptors consist of rods and cones that lie behind bipolar cells that connect rods, cones, and nerve ganglions. Rods detect peripheral objects, black and white, movement, and dim light objects; they are located to the periphery of the macula. Cones, concentrated in the center of the retina, detect color, fine details, and function best in brighter light conditions.
Retinitis pigmentosa develops because the gene mutations send incorrect messages to the photoreceptor cells and RPE, causing production of the wrong types and wrong amounts of protein that would normally allow the cells to function properly. Loss of rod photoreceptors is followed by loss of cone receptors. An eye doctor can diagnose the condition by seeing dark pigment deposits on the retina.
Types of Retinitis Pigmentosa
Retinitis pigmentosa is typically genetically related. Even simple RP is highly complicated. Each genetic type is caused by mutations in several or many different genes.
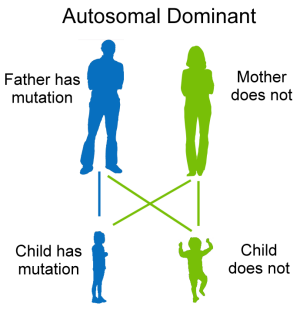 Dominant. In this form of genetic inheritance, an abnormal gene set from one parent can cause disease, even though the matching gene from the other parent is normal.
Dominant. In this form of genetic inheritance, an abnormal gene set from one parent can cause disease, even though the matching gene from the other parent is normal.
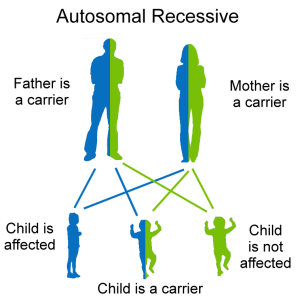 Recessive. In recessive inheritance, a copy from each parent of an abnormal gene must be present in order for the disease or trait to develop.
Recessive. In recessive inheritance, a copy from each parent of an abnormal gene must be present in order for the disease or trait to develop.
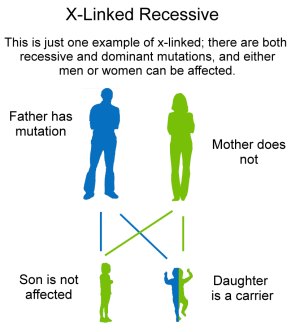 X-linked. X-linked recessive means that the mother carries the mutated gene, and her sons have a 50% chance of being affected.
X-linked. X-linked recessive means that the mother carries the mutated gene, and her sons have a 50% chance of being affected.
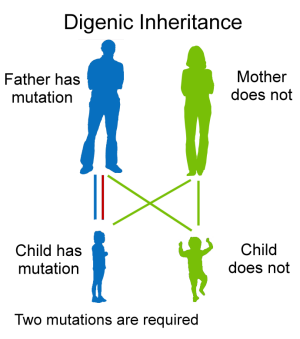 Rare mitochondrial and digenic forms. Digenic inheritance (DI) is the simplest form of inheritance for genetically complex diseases.
Rare mitochondrial and digenic forms. Digenic inheritance (DI) is the simplest form of inheritance for genetically complex diseases.
Symptoms
The typical symptoms include decreased night vision and peripheral vision, usually starting in the first or second decade of life. Marked loss of vision occurs over many years but may develop slowly. The diagnosis is made by a thorough eye examination and a special test called an electroretinogram, or "ERG".
- Poor adaption in adjusting from light to dark or dark to light.
- Poor night vision - difficulty in low light conditions
- Poor peripheral vision - gives the effect of tunnel vision
- Poor color detection - errors in color vision in both red-green and blue-yellow ranges
Causes
Gene mutations, dominant, recessive, or x-linked are carried from parent to child. Digenic inheritance, another form, requires two mutated genes. More than 100 different mutations have been identified of the gene-controlling rhodopsin, that support low light vision, and are associated with the variants of retinitis pigmentosa.
In other words, many different types of mutations of the gene controlling rhodopsin have been discovered. Rhodopsin allows our eyes to work in low-light conditions. Since the discovery of its link to retinitis pigmentosa in 1989, one hundred other mutations have been found that account for variations of retinal deterioration.
Environment. Mutations can occur during one’s lifetime, often related to environmental factors that include ultraviolet radiation from the sun, exposure to toxins, or possibly additives and chemicals added to our foods. Mutations may also occur if a mistake is made when DNA copies itself during cell division.
Related conditions
Retinitis pigmentosa usually affects the eye, but there are several other rare conditions that may be correlated.
- Usher syndrome wherein hearing is affected. The condition is associated with gene mutation.
- Leber's in which mitochondria, the energy producers of retinal nerve cells, malfunction.
- Bardet-Viedl syndrome is often associated with obesity present at childhood and other problems evident at childhood.
- Senior Loken syndrome and Alport syndrome are severe forms of retinitis pigmentosa associated with kidney problems
- Cohen syndrome, Cockayne syndrome and Jeune syndrome connect retinitis pigmentosa with body structure disorders.
- There are a number of diseases that associate retinitis pigmentosa with a wide variety of conditions involving metabolism of vitamins, proteins, lipids, etc.
- Other conditions associate RP with various neurological conditions.
Next:
Nutritional support,
diet, & lifestyle tips for retinitis pigmentosa.
Retinitis Pigmentosa News
Want to learn more? See our blog for news on pigmentosa.


{kind=link}