There are three types of Usher (USH), all with similar symptoms. Different genes have been identified for each type. When an eye doctor looks at the retina in a patient with Usher, the optic nerve looks very pale, the blood vessels supplying nutrients to the retina are thin, and the doctor can see that there are characteristic spots and splotches of pigment called bone spicules.
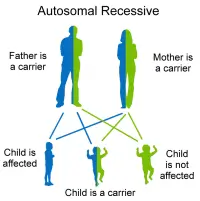
- Type 1 Usher. At birth, the child is severely deaf with severe balancing problems. Before age 10 years old, the child may begin to develop vision problems. This genetic disorder has been identified frequently in today’s Ashkenazi Jewish population. This type of Usher syndrome includes problems with the inner ear affecting balance that results in children being unable to sit or walk independently until later than usual.
- Type 2 Usher. At birth the child has moderate to serious deafness and normal capacity to balance. These children are assisted by a hearing aids. Retinitis pigmentosa isn’t noticed until they are in their teens. Type 2 is thought to be the most common form of Usher syndrome, although the frequency is unknown. The vision problems of this type 2 tend to progress more slowly than those of type 1.
- Type 3 Usher. These children do not have hearing problems at birth and they have normal or almost normal balance. However hearing and vision deteriorate with time, usually by the teen years. Balance difficulties might develop later on. Hearing aids are usually needed by adulthood, and night blindness may start in early teens. Blind spots can develop and the person is often legally blind by mid-adulthood. The clinical diagnosis refers to a central visual acuity of 20/200 or less in the better eye, with the best possible correction and/or a visual field of 20 degrees or less. Type 3 Usher syndrome accounts for only a small percentage of all Usher syndrome cases in most populations. This 3rd type is prevalent in Finland.
 Next: Nutrients, diet, & lifestyle for Usher syndromes.
Next: Nutrients, diet, & lifestyle for Usher syndromes.
Want to learn more? See our blog for news on photoreceptor dysfunction.
Next: Nutrients, diet, & lifestyle for Usher syndromes.